





GIỚI THIỆU:
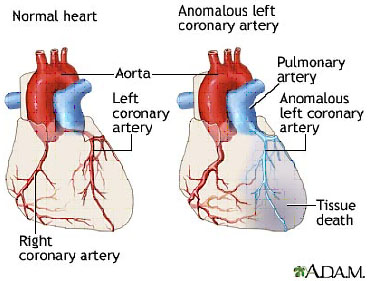
Hội chứng ALCAPA (The Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery) là một bất thường bẩm sinh hiếm gặp của động mạch vành (ĐMV), trong đó ĐMV trái xuất phát bất thường từ động mạch phổi thay vì từ động mạch chủ[1] (hình 1).
Hình 1
ALCAPA được mô tả đầu tiên vào năm 1866. Mô tả triệu chứng lâm sàng đầu tiên kết hợp với những đặc điểm trên sinh thiết tử thi được mô tả bởi Bland và cộng sự vào năm 1933, cho nên hội chứng này còn gọi là hội chứng Bland-White-Garland:
Thông thường trẻ khỏe chỉ một vài tháng sau khi trẻ bắt đầu có triệu chứng của bệnh tim. Thỉnh thoảng những bệnh nhân có thể không có triệu chứng ngay cả khi đến tuổi trưởng thành nhưng thường trẻ tử vong xảy ra trong giai đoạn còn nhỏ [1]. Khoảng 85% bệnh nhân ALCAPA chết khi còn nhỏ và ALCAPA gặp ở người lớn tuổi là rất hiếm [2].
Gần đây, tiên lượng của ALCAPA được cải thiện đáng kể khi được chẩn đoán sớm dựa vào sự kết hợp của các phương tiện chẩn đoán hình ảnh như siêu âm tim và MSCT và sự cải thiện của kỹ thuật mổ.
Ngày nay, MSCT đã trở thành một phương tiện chẩn đoán hình ảnh hữu dụng trong đánh giá động mạch vành không xâm nhập, đặc biệt MSCT xác định được vị trí xuất phát và đường đi của động mạch vành cũng như mối tương quan giữa động mạch vành với các cấu trúc lân cận nhờ vào hình ảnh 3 D.
MỤC TIÊU:
Đánh giá vai trò của MSCT trong chẩn đoán, điều trị cũng như tiên lượng hội chứng ALCAPA.
VẬT LIỆU VÀ PHƯƠNG PHÁP:
Có 14 trường hợp ALCAPA trong 5050 trường hợp được chụp MSCT- 64 động mạch vành tại MEDIC trong thời gian từ 9/2006 đến 4/2010.
KẾT QUẢ VÀ BÀN LUẬN:
Có 14 trường hợp ALCAPA trong 5050 trường hợp đến chụp MSCT -64 động mạch vành, chiếm tỷ lệ gần bằng 0.28%. Gồm 4 nam (28.6%), 10 nữ (71.4%), tuổi trung bình trong nhóm là 15 ±22 tuổi, tuổi lớn nhất là 67, nhỏ nhất là 3 tháng tuổi. Ở Mỹ tần suất bệnh xảy ra khoảng 1/300000 trẻ sinh ra và chiếm 0.25-0.5% trong tổng số bệnh tim bẩm sinh [1]. Khoảng 90% trường hợp ALCAPA đột tử ở tuổi trung bình là 35 tuổi và có vài bệnh nhân sống đến 50 tuổi mà không được điều trị phẩu thuật [2]. Trong y văn, có một trường hợp ALCAPA ở bệnh nhân nữ 72 tuổi không có triệu chứng lâm sàng mặc dù có luồng thông trái phải rõ rệt [2].
Hệ tuần hoàn động mạch vành được thành lập vào ngày thứ 45 của thai kỳ. ALCAPA được gây ra do sự phân chia bất thường của thân chóp hoặc bởi sự phân chia bất thường của chồi nội mạc mà hiện diện trên 6 xoang valsalva của đại động mạch. Thông thường 2 chồi nội mạc trong xoang valsalva của động mạch chủ trở thành ĐMV. Trong trường hợp ALCAPA, một chồi nội mạc thỉnh thoảng tồn tại trên xoang động mạch phổi (ĐMP) và phát triển thành thân chung ĐMV trái. Ngoài ra, ĐMV trái có thể nối đến vị trí khác trên ĐMP và đã trường hợp được báo cáo là nối đến một nhánh của ĐMP. Thông thường ALCAPA xảy ra đơn độc không kèm bất thường khác, tuy nhiên đôi khi ALCAPA phối hợp với những bất thường bẩm sinh khác bao gồm: thông liên thất, còn ống động mạch, hẹp eo động mạch chủ [1]. Trong 14 trường hợp ALCAPA của chúng tôi có một trường hợp ALCAPA kèm theo còn ống động mạch (Hình 2).
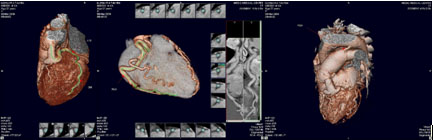
Hình 2: Trường hợp ALCAPA ở bệnh nhân nữ 27 tuổi, được chẩn đoán dò ĐMV trái vào ĐMP trên siêu âm tim trước khi chụp MSCT-64 động mạch vành, ở bệnh nhân này ngoài ALCAPA còn có còn ống động mạch đi kèm. Bệnh nhân này đã được phẩu thuật tại bệnh viện Chợ Rẫy.
Theo timmachhoc.vn
PK Đức Tín
Tin tức liên quan
Điện thoại bàn: (028) 3981 2678
Di động: 0903 839 878 - 0909 384 389