





Đột tử một vấn đề lớn của y học. Chúng ta đã biết về một bản tổng hợp lâm sàng hàng đầu về các nguyên nhân gây đột tử do tim (SCD). Từ các khu vực địa lý khác nhau trên thế giới xuất hiện quan điểm lâm sàng khác nhau đối với vấn đề SCD và làm sáng tỏ các yếu tố dự đoán và ước tính tỷ lệ lưu hành trong di truyền trên toàn thế giới.
CÁC HỘI CHỨNG RỐI LOẠN NHỊP KẾT HỢP VỚI ĐỘT TỬ TIM
TÓM TẮT
Đột tử (Sudden death: SD) được định nghĩa là tử vong tự nhiên không thể đoán trước của một cá nhân khỏe mạnh. Nguyên nhân chính của SD là do các biến đổi của tim, và do đó được gọi là đột tử do tim (Sudden cardiac death: SCD). Những tiến bộ gần đây trong khoa tim mạch đã phát triển các hướng dẫn mới trong chẩn đoán, điều trị cũng như phòng ngừa các bệnh liên quan đến SCD. Mặc dù thực tế này, việc xác định sớm các cá nhân có nguy cơ vẫn là một trong những thách thức chính hiện nay đối với các nhà tim mạch học. Một nguyên nhân chính của SCD là những biến đổi tố bẩm và các bệnh tim cấu trúc mặc dù một số lượng đáng kể không cho thấy bất kỳ khiếm khuyết tim nào trong quá trình khám nghiệm tử thi. Trong những trường hợp chưa được giải quyết này, bệnh kênh ion được coi là nguyên nhân tiềm năng đầu tiên của SD. Do tất cả các bệnh này có nguồn gốc di truyền, các thành viên gia đình có thể có nguy cơ, mặc dù không có triệu chứng. Thật không may, SCD thường là biểu hiện lâm sàng đầu tiên và duy nhất của một bệnh tim di truyền vẫn không bị phát hiện bằng các nghiên cứu lâm sàng thông thường. Trong một số trường hợp, hoạt động thể chất có thể là tác nhân kích hoạt SCD như là triệu chứng đầu tiên. Những tiến bộ công nghệ gần đây trong di truyền học đã cải thiện việc sử dụng xét nghiệm di truyền vào lĩnh vực SCD. Nó có thể giúp xác định nguyên nhân tử vong ở những bệnh nhân bị ảnh hưởng, trong các trường hợp sau khi chết mà không có chẩn đoán kết luận cũng như các thành viên gia đình không có triệu chứng có nguy cơ. Phần này tập trung vào những tiến bộ gần đây về rối loạn nhịp tim liên quan đến SCD.
MỞ ĐẦU
Đột tử (SD) được định nghĩa là một biến cố bất ngờ và tự nhiên xảy ra trong vòng một giờ đầu tiên kể từ khi xuất hiện các triệu chứng ở một người khỏe mạnh và trong đó khám nghiệm tử thi kỹ lưỡng không chứng minh được nguyên nhân tử vong (Basso et al., 2010). Do đó, nó đại diện cho một gánh nặng to lớn đối với gia đình, cộng đồng và chăm sóc sức khỏe, đặc biệt là khi xảy ra ở trẻ em. Biến đổi tim là nguyên nhân chính của SD (SCD). Ở quần thể dân số > 50 tuổi, gần 80% trường hợp SCD là hậu quả của bệnh tim mạch vành, trong khi ở dân số < 35 tuổi, SCD chủ yếu là do biến đổi di truyền (Oliva et al., 2010). Với bản chất di truyền của nó, có ý nghĩa quan trọng đối với các thành viên gia đình. Do đó, xét nghiệm di truyền có mục tiêu để tăng hiệu suất chẩn đoán, tạo điều kiện sàng lọc di truyền theo tầng của các thành viên gia đình với xét nghiệm tập trung hơn (Campuzano et al., 2014b). Một hạn chế lớn trong chẩn đoán lâm sàng của các gia đình là sự xâm nhập không đầy đủ và biểu hiện thay đổi, cản trở việc xác định tất cả người thân có nguy cơ mắc SCD (Giudicessi và Ackerman, 2013). Trong một số trường hợp, hoạt động thể chất có thể là tác nhân kích hoạt SCD như là triệu chứng đầu tiên. Tập trung vào các hội chứng di truyền, hai nhóm chính đã được đưa ra: bệnh lý kênh ion, trong đó chất nền gây rối loạn nhịp tim được tìm thấy trong sự bất thường của các tính chất điện của tim mà không có bất kỳ khiếm khuyết cấu trúc nào, và bệnh cơ tim, trong đó rối loạn nhịp tim có liên quan đến sự hiện diện của tim thay đổi cấu trúc tim (Wellens et al., 2014). Nghiên cứu di truyền đã chỉ ra các hội chứng di truyền liên quan đến SCD là do sự thay đổi bệnh lý trong gen mã hóa bốn loại protein của tế bào cơ: sarcomeric, cytoskeletal, desmosomal và các kênh ion hoặc protein liên quan. Trong khi chất nền cơ học là khác nhau ở cả hai nhóm, điểm kết thúc là một đặc điểm chung (Campuzano et al., 2014a). Trong phần này, chúng tôi sẽ tập trung vào cơ sở di truyền của rối loạn nhịp tim liên quan đến SCD.
BỆNH KÊNH ION
Như đã đề cập ở trên, thuật ngữ “bệnh kênh ion”, đề cập đến bệnh tim được đặc trưng bằng tim bình thường về cấu trúc dẫn đến rối loạn nhịp tim, ngất và SCD (Martin et al., 2013). Những bệnh rối loạn nhịp tim này được gây ra do các biến thể bệnh lý chủ yếu ở các gen mã hóa các kênh ion tim (chủ yếu là natri, kali và canxi) hoặc các protein liên quan. Nói chung, tùy thuộc vào kênh ion nào bị ảnh hưởng, các hội chứng khác nhau sẽ biểu hiện. Tuy nhiên, cùng một hội chứng có thể cho thấy một mức độ chồng chéo nhất định nếu các loại kênh khác nhau có thể bị ảnh hưởng.
Bảng 1. Các gene kết hợp với các bệnh kênh ion
| Kênh | Bệnh | Di truyền | GENE (ID) |
|
Natri |
LQT |
Nhiễm sắc thể thường trội | SCN5A (6331)
SCN4B (6330) SCN1B (6324) |
| BrS | Nhiễm sắc thể thường trội | SCN5A (6331)
GPD1L (23171) SCN1B (6324) SCN3B (55800) SCN2B (6327) SCN10A (6336) |
|
|
Natri được kết hợp |
LQT | Nhiễm sắc thể thường trội | CAV3 (859)
SNTA1 (6640) |
|
BrS |
Nhiễm sắc thể thường trội |
RANGRF (29098) SLMAP (7871)
PKP2 (5318) |
|
|
Kali |
LQT |
Nhiễm sắc thể thường trội |
KCNQ1 (3784)
KCNH2 (3757) KCNE1 (3753) KCNE2 (9992) KCNJ5 (3762) KCNJ2 (3759) |
| Nhiễm sắc thể thường lặn | KCNQ1 (3784)
KCNE1 (3753) |
||
| SQT |
Nhiễm sắc thể thường trội |
KCNH2 (3757)
KCNQ1 (3784) KCNJ2 (3759) |
|
|
BrS |
Nhiễm sắc thể thường trội |
ABCC9 (10060)
KCNE3 (10008) KCNJ8 (3764) HCN4 (10021) KCND3 (3752) |
|
| Trội liên quan giới tính | KCNE5 (23630) | ||
| CPVT | Nhiễm sắc thể thường trội | KCNJ2 (3759) | |
| Kali được kết hợp | LQT | Nhiễm sắc thể thường trội | AKAP9 (10142) |
|
Canxi |
BrS |
Nhiễm sắc thể thường trội |
CACNA1C (775) CACNB2 (785) CACNA2D1 (781) TRPM4 (54795) |
| SQT | Nhiễm sắc thể thường trội | CACNA2D1 (781) | |
| LQT | Nhiễm sắc thể thường trội | CACNA1C (775) RYR2 (6262) CALM1 (801) CALM2 (805) | |
| CPVT | Nhiễm sắc thể thường trội | RYR2 (6262) | |
| Nhiễm sắc thể thường lặn | CASQ2 (845) | ||
|
Canxi được kết hợp |
LQT | Nhiễm sắc thể thường trội | ANK2 (284) |
| Nhiễm sắc thể thường lặn | TRDN (10345) | ||
| CPVT | Nhiễm sắc thể thường trội
Nhiễm sắc thể thường trội |
CALM1 (801)
CALM2 (805) |
|
| Nhiễm sắc thể thường lặn | TRDN (10345) |
BrS: Hội chứng Brugada, CPVT: Nhịp nhanh thất đa hình liên quan với catecholamine, LQTS: Hội chứng QT dài. SQTS: Hội chứng QT ngắn.
Ngoài ra, nó còn được biết rõ sự tương tác của di truyền và môi trường như là một công cụ sửa đổi kiểu hình (Campuzano et al., 2010). Các kênh ion là glycoprotein được gắn trong màng tế bào cơ cho phép dòng ion vào và ra khỏi tế bào để điều chỉnh sự chênh lệc điện học. Dòng ion di chuyển điện tích và gây ra sự hình thành điện thế hoạt động của tim (dòng điện), được điều chỉnh bằng cách mở và đóng đồng bộ các kênh theo độ chênh điện thế. Những bệnh này thường ảnh hưởng đến việc tạo và truyền điện qua tế bào cơ, một bước quan trọng trong hoạt động cơ điện, đòi hỏi sự phối hợp của một số nguyên tố, bao gồm các kênh ion và protein cấu trúc, được đặt tên là phức hợp đại phân tử kênh ion (Martin et al., 2013). Sự phức tạp của quá trình này vẫn là một hạn chế lớn để hiểu các cơ chế gây rối loạn nhịp tim.
Hiện nay, các bệnh kênh chính là: hội chứng Brugada (BrS), hội chứng QT dài (LQTS), hội chứng QT ngắn (SQTS) và nhịp nhanh thất đa hình liên quan catecholamine (CPVT) (Priori và BlomstromLundqvist, 2015). Chúng ta sẽ tập trung vào 4 hội chứng rối loạn nhịp tim:
- Hội chứng Brugada
Hội chứng Brugada (BrS) là một thực thể hiếm gặp, được đặc trưng bằng sự chênh lên của đoạn ST type vòm (coved-type STsegment elevation) trong block nhánh bó phải không điển hình ở chuyển đạo V1 đến V3 của ECG, không có bệnh tim cấu trúc (Brugada và Brugada, 1992). Mẫu ECG có thể là cơ bản hoặc không liên tục, và nó có thể được bộc lộ trong quá trình test thuốc (thuốc chẹn kênh natri nhóm IC). Tỷ lệ hiện mắc được ước tính là 3-5 trên 10000 người mỗi năm và độ tuổi trung bình khởi phát các biến cố là khoảng 40 tuổi, nam giới bị ảnh hưởng là chính (75%). SCD liên quan đến BrS thường xảy ra trong khi ngủ, khi nghỉ ngơi hoặc khi tăng trương lực phế vị (Brugada et al., 2014). Bệnh nhân mắc BrS thường vẫn không có triệu chứng và các yếu tố điều biến như sốt, gắng sức hoặc thuốc (www.brugadadrugs.org), có thể đóng vai trò chính trong đặc tính động học của ECG. Mặc dù SCD trong hội chứng này không phổ biến trong khi gắng sức, khuyến cáo cho các vận động viên cạnh tranh với bất thường về tim mạch, khuyến cáo bệnh nhân BrS nên tránh tập thể dục cường độ cao. Việc quản lý bệnh nhân được chẩn đoán BrS là khó khăn vì thiếu liệu pháp y tế hiệu quả để ngăn ngừa rối loạn nhịp tim. Do đó, các kế hoạch phân tầng nguy cơ khá hạn chế vì ít được biết về biểu hiện lâm sàng và tiên lượng. Ở những bệnh nhân có chẩn đoán BrS và tiền sử ngất, SD được cứu sống, thở ngắc ngoải về ban đêm (nocturnal agonal breathing) hoặc co giật về đêm, được coi là một nhóm bệnh nhân có nguy cơ SCD cao hơn, và cấy ICD được chỉ định. Hiện tại, ICD là biện pháp phòng ngừa tốt nhất trong BrS. Tuy nhiên, cấy ICD khi có triệu chứng không phải là không có tranh cãi, đặc biệt là ở trẻ em (Sarquella-Brugada et al., 2016). Quyết định cấy ICD ở trẻ em không phải là một công việc dễ dàng vì đây là một liệu pháp suốt đời, không thể không bị biến chứng.
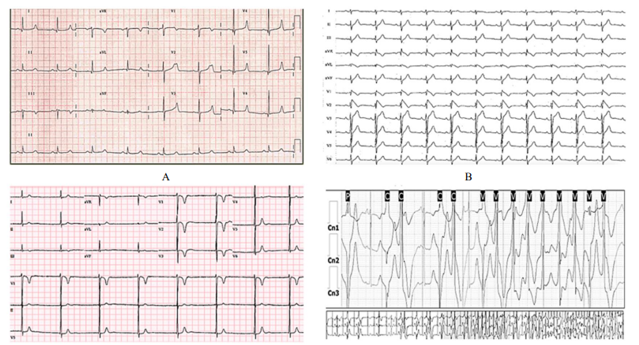
Hình 1. Điện tâm đồ cho thấy A. Hội chứng QT dài. B. Hội chứng Brugada. C. Hội chứng QT ngắn. D. Nhịp nhanh thất đa hình liên quan catecholamine.
Có dữ liệu hạn chế chứng minh lợi ích của các liệu pháp phòng ngừa khác nhưng nếu bệnh nhân cũng có biểu hiện rối loạn nhịp tim, Quinidine có thể được chỉ định khi bệnh nhân đã được cấy ICD, gây ra bão điện. BrS có thể liên kết rối loạn dẫn truyền AV và nhịp nhanh trên thất, vì vậy chúng ta đặt câu hỏi về sự có mặt của đánh trống ngực và cuối cùng điều trị các rối loạn nhịp tim này bằng triệt phá bằng năng lượng tần số radio qua catheter có thể được xem xét (Arbelo và Brugada, 2014).
Liên quan đến các nền tảng di truyền, cho đến nay hơn 250 biến thể gây bệnh đã được xác định trong 19 gen: [ABCC9, CACNA1C, CACNA2D1, CACNB2b, GPD1-L, HCN4, KCND3, KCNE3, KCNE5, KCNJ8, KCN, SCN3B, SCN5A, SLMAP và TRPM4] (Sarquella-Brugada và cộng sự., 2016). Một phân tích di truyền toàn diện chỉ xác định nguyên nhân gây bệnh ở 35% -40% gia đình và khoảng 25% – 30% trong số những bệnh nhân dương tính này có biến thể gây bệnh ở SCN5A (BrS type 1). Do đó, các hướng dẫn hiện tại chỉ đề xuất phân tích di truyền của gen này (Priori et al., 2015). Gen SCN5A mã hóa kênh natri tim Nav1.5 và các biến thể gây bệnh gây mất chức năng (Chen và cộng sự., 1998). Các biến thể gây bệnh khác đã được công bố trong các tiểu đơn vị beta làm thay đổi Nav1.5 (tăng hoặc giảm INa) và được mã hóa bằng SCN1B, SCN2B và SCN3B (Hu và cộng sự., 2009; Riuro và cộng sự., 2013; Watanabe và cộng sự., 2008). Gần đây, người ta đã công bố ngụ ý của SCN10A, một gen mã hóa kênh natri thần kinh Nav1.8 điều chỉnh biểu lộ Nav1.5 (Bezzina và cộng sự., 2013), nhưng một số nghiên cứu cần được thực hiện để làm rõ vai trò của gen này trong BrS. Ngoài ra, các biến thể gây bệnh trong gen GPD1-L làm giảm cả biểu hiện màng bề mặt và dòng natri vào bên trong (London cộng sự., 2007). Gần đây, gen RANGRF (protein MOG1) đã được báo cáo làm suy yếu việc trao đổi Nav1.5 đến màng (Kattygnarath cộng sự., 2011). Vào năm 2012, một biến thể gây bệnh đã được xác định trong SLMAP, một gen mã hóa protein liên quan đến màng sarcolemmal, được định vị tại các ống T và mạng lưới sarcoplasmic và điều chỉnh việc trao đổi nội bào của kênh hNav1.5 (Ishikawa cộng sự., 2012). Gần đây, người ta cũng đã báo cáo các biến thể gây bệnh trong PKP2 (protein plakophilin-2). Đây là một gen desmosomal và mối tương quan giữa sự mất biểu hiện của plakophilin-2 và Ina giảm (Cerrone và Delmar, 2014; Cerrone và cộng sự., 2014). Liên quan đến các kênh kali, bằng chứng đầu tiên cho thấy biến thể gây bệnh mới có chức năng gây bệnh trong KCND3 liên quan đến BrS đã được công bố vào năm 2011 (Giudicessi và cộng sự., 2011). Gen này mã hóa một thành phần của kênh kali, bị kiểm soát điện áp và nổi trội trong giai đoạn tái cực của điện thế hoạt động. Những thay đổi di truyền khác đã được báo cáo trong gen KCNE3 (protein MiRPS2) mã hóa một tiểu đơn vị điều tiết của kênh kali dưới đơn vị β của kênh Ito truyền kali ra ngoài (Delpon và cộng sự., 2008). Mặc dù BrS chủ yếu theo mô hình di truyền trội tự phát, một biến thể gây bệnh liên quan đến BrS đã được định vị trong gen KCNE1L (KCNE5) – X-link- (Ohno cộng sự., 2011). Gần đây, một gia đình BrS mang biến thể gây bệnh trong gen KCNJ8 cũng đã được báo cáo (Medeiros-Domingo và cộng sự, 2010). Ngoài ra, BrS cũng được liên kết với HCN4 mã hóa kênh 4 kali qua cổng nucleotide vòng được hoạt hóa phân cực quá mức (Ueda và cộng sự., 2009). Nó được biểu hiện ở nút xoang và các tế bào của hệ thống dẫn truyền tim, và mất chức năng của protein này có liên quan đến rối loạn chức năng nút xoang. Gen ABCC9 mã hóa thụ thể sulfonylurea tiểu đơn vị SUR2A. Các biến thể gây bệnh trong gen này gây ra sự tăng chức năng trong I (K-ATP) khi kết hợp với mất chức năng trong SCN5A và có thể có kiểu hình rối loạn nhịp tim nghiêm trọng (Hu và cộng sự., 2014). Cuối cùng, sự thay đổi di truyền trong các kênh canxi hoặc protein liên quan cũng đã được báo cáo trong các gia đình BrS. Các biến thể di truyền trong CACNA1C chịu trách nhiệm cho một đơn vị kênh canxi loại L bị khiếm khuyết. Điều đó gây ra mất chức năng kênh, nhưng được liên kết với khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Các biến thể di truyền khác trong CACNB2b dẫn đến cùng dấu vết ECG cho thấy sự kết hợp giữa BrS và khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Năm 2010, gen CACNA2D1 được liên kết với BrS (Burashnikov và cộng sự., 2010). Cuối cùng, các biến thể di truyền gây bệnh trong gen TRPM4 cũng đã được báo cáo. Gen này mã hóa protein melastatin điện thế thụ cảm thể tạm thời số 4, kênh cation không chọn lọc được canxi hoạt hóa, và là thành viên của gia đình lớn của các gen điện thế thụ thể tạm thời (Stallmeyer et al., 2012).
Theo timmachhoc
PK Đức Tín
Tin tức liên quan
Điện thoại bàn: (028) 3981 2678
Di động: 0903 839 878 - 0909 384 389