





Đột tử một vấn đề lớn của y học. Chúng ta đã biết về một bản tổng hợp lâm sàng hàng đầu về các nguyên nhân gây đột tử do tim (SCD). Từ các khu vực địa lý khác nhau trên thế giới xuất hiện quan điểm lâm sàng khác nhau đối với vấn đề SCD và làm sáng tỏ các yếu tố dự đoán và ước tính tỷ lệ lưu hành trong di truyền trên toàn thế giới.
- Hội chứng Brugada
Hội chứng Brugada (BrS) là một thực thể hiếm gặp, được đặc trưng bằng sự chênh lên của đoạn ST type vòm (coved-type STsegment elevation) trong block nhánh bó phải không điển hình ở chuyển đạo V1 đến V3 của ECG, không có bệnh tim cấu trúc (Brugada và Brugada, 1992). Mẫu ECG có thể là cơ bản hoặc không liên tục, và nó có thể được bộc lộ trong quá trình test thuốc (thuốc chẹn kênh natri nhóm IC). Tỷ lệ hiện mắc được ước tính là 3-5 trên 10000 người mỗi năm và độ tuổi trung bình khởi phát các biến cố là khoảng 40 tuổi, nam giới bị ảnh hưởng là chính (75%). SCD liên quan đến BrS thường xảy ra trong khi ngủ, khi nghỉ ngơi hoặc khi tăng trương lực phế vị (Brugada et al., 2014). Bệnh nhân mắc BrS thường vẫn không có triệu chứng và các yếu tố điều biến như sốt, gắng sức hoặc thuốc (www.brugadadrugs.org), có thể đóng vai trò chính trong đặc tính động học của ECG. Mặc dù SCD trong hội chứng này không phổ biến trong khi gắng sức, khuyến cáo cho các vận động viên cạnh tranh với bất thường về tim mạch, khuyến cáo bệnh nhân BrS nên tránh tập thể dục cường độ cao. Việc quản lý bệnh nhân được chẩn đoán BrS là khó khăn vì thiếu liệu pháp y tế hiệu quả để ngăn ngừa rối loạn nhịp tim. Do đó, các kế hoạch phân tầng nguy cơ khá hạn chế vì ít được biết về biểu hiện lâm sàng và tiên lượng. Ở những bệnh nhân có chẩn đoán BrS và tiền sử ngất, SD được cứu sống, thở ngắc ngoải về ban đêm (nocturnal agonal breathing) hoặc co giật về đêm, được coi là một nhóm bệnh nhân có nguy cơ SCD cao hơn, và cấy ICD được chỉ định. Hiện tại, ICD là biện pháp phòng ngừa tốt nhất trong BrS. Tuy nhiên, cấy ICD khi có triệu chứng không phải là không có tranh cãi, đặc biệt là ở trẻ em (Sarquella-Brugada et al., 2016). Quyết định cấy ICD ở trẻ em không phải là một công việc dễ dàng vì đây là một liệu pháp suốt đời, không thể không bị biến chứng.
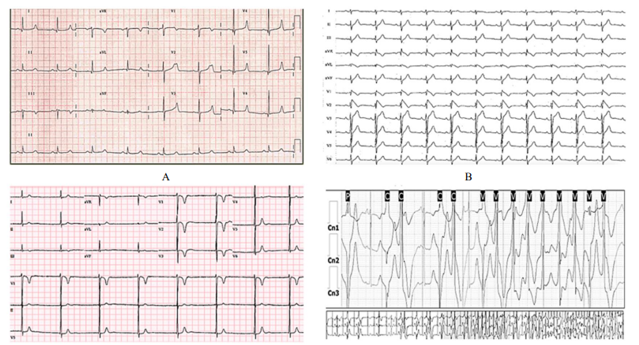
Hình 1. Điện tâm đồ cho thấy A. Hội chứng QT dài. B. Hội chứng Brugada. C. Hội chứng QT ngắn. D. Nhịp nhanh thất đa hình liên quan catecholamine.
Có dữ liệu hạn chế chứng minh lợi ích của các liệu pháp phòng ngừa khác nhưng nếu bệnh nhân cũng có biểu hiện rối loạn nhịp tim, Quinidine có thể được chỉ định khi bệnh nhân đã được cấy ICD, gây ra bão điện. BrS có thể liên kết rối loạn dẫn truyền AV và nhịp nhanh trên thất, vì vậy chúng ta đặt câu hỏi về sự có mặt của đánh trống ngực và cuối cùng điều trị các rối loạn nhịp tim này bằng triệt phá bằng năng lượng tần số radio qua catheter có thể được xem xét (Arbelo và Brugada, 2014).
Liên quan đến các nền tảng di truyền, cho đến nay hơn 250 biến thể gây bệnh đã được xác định trong 19 gen: [ABCC9, CACNA1C, CACNA2D1, CACNB2b, GPD1-L, HCN4, KCND3, KCNE3, KCNE5, KCNJ8, KCN, SCN3B, SCN5A, SLMAP và TRPM4] (Sarquella-Brugada và cộng sự., 2016). Một phân tích di truyền toàn diện chỉ xác định nguyên nhân gây bệnh ở 35% -40% gia đình và khoảng 25% – 30% trong số những bệnh nhân dương tính này có biến thể gây bệnh ở SCN5A (BrS type 1). Do đó, các hướng dẫn hiện tại chỉ đề xuất phân tích di truyền của gen này (Priori et al., 2015). Gen SCN5A mã hóa kênh natri tim Nav1.5 và các biến thể gây bệnh gây mất chức năng (Chen và cộng sự., 1998). Các biến thể gây bệnh khác đã được công bố trong các tiểu đơn vị beta làm thay đổi Nav1.5 (tăng hoặc giảm INa) và được mã hóa bằng SCN1B, SCN2B và SCN3B (Hu và cộng sự., 2009; Riuro và cộng sự., 2013; Watanabe và cộng sự., 2008). Gần đây, người ta đã công bố ngụ ý của SCN10A, một gen mã hóa kênh natri thần kinh Nav1.8 điều chỉnh biểu lộ Nav1.5 (Bezzina và cộng sự., 2013), nhưng một số nghiên cứu cần được thực hiện để làm rõ vai trò của gen này trong BrS. Ngoài ra, các biến thể gây bệnh trong gen GPD1-L làm giảm cả biểu hiện màng bề mặt và dòng natri vào bên trong (London cộng sự., 2007). Gần đây, gen RANGRF (protein MOG1) đã được báo cáo làm suy yếu việc trao đổi Nav1.5 đến màng (Kattygnarath cộng sự., 2011). Vào năm 2012, một biến thể gây bệnh đã được xác định trong SLMAP, một gen mã hóa protein liên quan đến màng sarcolemmal, được định vị tại các ống T và mạng lưới sarcoplasmic và điều chỉnh việc trao đổi nội bào của kênh hNav1.5 (Ishikawa cộng sự., 2012). Gần đây, người ta cũng đã báo cáo các biến thể gây bệnh trong PKP2 (protein plakophilin-2). Đây là một gen desmosomal và mối tương quan giữa sự mất biểu hiện của plakophilin-2 và Ina giảm (Cerrone và Delmar, 2014; Cerrone và cộng sự., 2014). Liên quan đến các kênh kali, bằng chứng đầu tiên cho thấy biến thể gây bệnh mới có chức năng gây bệnh trong KCND3 liên quan đến BrS đã được công bố vào năm 2011 (Giudicessi và cộng sự., 2011). Gen này mã hóa một thành phần của kênh kali, bị kiểm soát điện áp và nổi trội trong giai đoạn tái cực của điện thế hoạt động. Những thay đổi di truyền khác đã được báo cáo trong gen KCNE3 (protein MiRPS2) mã hóa một tiểu đơn vị điều tiết của kênh kali dưới đơn vị β của kênh Ito truyền kali ra ngoài (Delpon và cộng sự., 2008). Mặc dù BrS chủ yếu theo mô hình di truyền trội tự phát, một biến thể gây bệnh liên quan đến BrS đã được định vị trong gen KCNE1L (KCNE5) – X-link- (Ohno cộng sự., 2011). Gần đây, một gia đình BrS mang biến thể gây bệnh trong gen KCNJ8 cũng đã được báo cáo (Medeiros-Domingo và cộng sự, 2010). Ngoài ra, BrS cũng được liên kết với HCN4 mã hóa kênh 4 kali qua cổng nucleotide vòng được hoạt hóa phân cực quá mức (Ueda và cộng sự., 2009). Nó được biểu hiện ở nút xoang và các tế bào của hệ thống dẫn truyền tim, và mất chức năng của protein này có liên quan đến rối loạn chức năng nút xoang. Gen ABCC9 mã hóa thụ thể sulfonylurea tiểu đơn vị SUR2A. Các biến thể gây bệnh trong gen này gây ra sự tăng chức năng trong I (K-ATP) khi kết hợp với mất chức năng trong SCN5A và có thể có kiểu hình rối loạn nhịp tim nghiêm trọng (Hu và cộng sự., 2014). Cuối cùng, sự thay đổi di truyền trong các kênh canxi hoặc protein liên quan cũng đã được báo cáo trong các gia đình BrS. Các biến thể di truyền trong CACNA1C chịu trách nhiệm cho một đơn vị kênh canxi loại L bị khiếm khuyết. Điều đó gây ra mất chức năng kênh, nhưng được liên kết với khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Các biến thể di truyền khác trong CACNB2b dẫn đến cùng dấu vết ECG cho thấy sự kết hợp giữa BrS và khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Năm 2010, gen CACNA2D1 được liên kết với BrS (Burashnikov và cộng sự., 2010). Cuối cùng, các biến thể di truyền gây bệnh trong gen TRPM4 cũng đã được báo cáo. Gen này mã hóa protein melastatin điện thế thụ cảm thể tạm thời số 4, kênh cation không chọn lọc được canxi hoạt hóa, và là thành viên của gia đình lớn của các gen điện thế thụ thể tạm thời (Stallmeyer et al., 2012).
- Hội chứng QT dài
Hội chứng QT dài (Long QT syndrome: LQTS) là một bệnh kênh tim không đồng nhất hiếm gặp, đặc trưng bằng sự kéo dài khoảng QT trên ECG (QTc> 460 ms nữ và> 450 ms nam) (Amin và cộng sự., 2013). Phổ của bất thường ECG gây ra sự mất ổn định điện bao gồm sóng T chẻ đôi hoặc hai pha và sóng T thay đổi luân phiên. Biểu hiện của LQTS thay đổi từ bệnh nhân không có triệu chứng đến các cơn ngất do nhịp nhanh thất (Torsade de Pointes) trong trạng thái của một tim bình thường về cấu trúc. LQTS có tỷ lệ mắc 1/2500 cá thể, là nguyên nhân chính gây SCD ở những người trẻ tuổi (Mizusawa và cộng sự, 2014). Ở tất cả các bệnh nhân, việc sử dụng beta-blocker ở liều cao rất được khuyến khích vì việc điều trị này làm giảm nguy cơ SCD mặc dù không cung cấp sự bảo vệ đầy đủ. Liều được điều chỉnh theo sự dung nạp với các thuốc này (www.torades.org). Tuy nhiên, tranh cãi tồn tại liên quan đến hiệu quả của thuốc chẹn beta chọn lọc tim, chẳng hạn như atenolol. Cấy ghép ICD là bắt buộc đối với những bệnh nhân bị SCD được cứu sống và những người có nguy cơ rối loạn nhịp tim gây tử vong (Behere và cộng sự., 2014). LQTS có thể là bẩm sinh hoặc mắc phải, nguyên nhân cuối cùng thường liên quan đến thuốc và mất cân bằng điện giải (hạ kali máu, hạ canxi máu và hạ kali máu). Dạng bẩm sinh có liên quan đến sự biến đổi bệnh lý trong các kênh ion và / hoặc các protein liên quan. Về di truyền học, LQTS có thể theo mô hình di truyền trội hoặc lặn tự phát. Hiện tại, hơn 1000 biến thể gây bệnh đã được xác định trong 18 gen (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, RYR2, SC1, SC1, SC). Tất cả các gen này cùng nhau chịu trách nhiệm cho 80% 85% của tất cả các trường hợp LQT (Nakano và Shimizu, 2016). Gần đây, việc sắp xếp lại bên trong (xóa và sao chép lớn) có liên quan đến LQTS, cho thấy nguyên nhân gây bệnh có thể là do các biến thể số sao chép (copy number variants: CNV) ảnh hưởng đến bất kỳ trong số 18 gen này. Tỷ lệ phát hiện CNV trong các gia đình LQTS dường như khoảng 2% -11%. Các gen chính liên quan đến LQTS là KCNQ1 – type 1- (30% -35%), KCNH2 -type 2- (25% -30%) và SCN5A type 3- (5% -10%) chịu trách nhiệm cho 65% – 75% của tất cả các trường hợp LQTS. Các hướng dẫn gần đây khuyên cáo chỉ nên thử nghiệm 3 gen chính này trong các gia đình được chẩn đoán mắc LQTS, đặc biệt là nếu có SCD của một thành viên trẻ trong gia đình (Priori và cộng sự., 2015). Các biến thể gây bệnh trong gen KCNQ1 chịu trách nhiệm chính gây ra khoảng QT kéo dài. Protein này liên kết với protein được mã hóa bằng gen KCNE1 (minK) để tạo thành phức hợp chức năng Iks (Bianchi et al., 1999). Các biến thể gây bệnh trong SCN5A gây ra khiếm khuyết chức năng dựa trên sự bất hoạt không hoàn toàn của kênh, do đó cho phép tiếp tục xâm nhập các ion natri vào tế bào trong quá trình tái cực và dẫn đến chức năng được gia tăng. Một gen liên quan đến bệnh khác là KCNH2 mã hóa tiểu đơn vị alpha của phức hợp Ikr (Sanguinetti et al., 1996); tiểu đơn vị alpha được xác định bằng gen KCNE2. Phức hợp Ikr này là tác nhân quan trọng nhất của quá trình tái cực nhanh trong giai đoạn 3. Các biến thể gây bệnh trong KCNH2 dẫn đến mất chức năng trong kênh Ikr và chiếm 35% đến 45% các trường hợp hội chứng LQT. Trong KCNE2, các biến thể gây bệnh cũng dẫn đến mất chức năng kênh. Một gen khác có liên quan đến hội chứng QT dài là KCNJ2 mã hóa protein Ik1 (Kir2.1) (hội chứng Tawil Anderson) (Tsuboi và Antzelevitch, 2006). Một gen kali khác liên quan đến hội chứng LQT là KCNJ5, mã hóa cho kênh Kir3.4 (còn được gọi là GIRK4) (Yang và cộng sự., 2010). Nó tạo thành các kênh đẳng hiệu hoặc các kênh không đồng nhất chức năng với Kir3.x khác, các kênh chịu trách nhiệm cho dòng protein kênh G được điều chỉnh bên trong (IKACh) và chủ yếu được biểu hiện ở nút xoang nhĩ, nút xoang thất và nhĩ. Một protein liên quan đến kali với LQT là protein neo A-kinase 9 (AKAPs), là protein tạo giàn xác định khu vực protein kinase A (PKA) và các protein khác điều chỉnh PKA (phosphatase hoặc các kinase khác) (Chen và cộng sự., 2007). Nó được mã hóa bằng gen AKAP9. Gen natri khác là SCN4B mã hóa tiểu đơn vị beta (NaVb4) của kênh natri. Tiểu đơn vị NaVb4 gây ra thay đổi âm tính về điện thế phụ thuộc natri trong kênh kích hoạt (MedeirosDomingo et al., 2007). Gần đây, một biến thể bệnh lý đã được xác định trong gen SCN1B chịu trách nhiệm về hội chứng LQT. Nó mã hóa cho hai đồng phân tim của tiểu đơn vị Navβ1: Navβ1 isoform a và Navβ1 isoform b (Riuro et al., 2014). Tập trung vào các protein liên quan đến natri, CAV3 mã hóa cho caveolin-3, đây là protein chính hình thành nên các khoảng trống trong cơ tim và cơ xương (Vatta et al., 2006). Protein kết hợp với natri khác là α1-Syntrophin, được mã hóa bằng gen SNTA1, một thành phần của protein liên quan đến loạn sản có chứa nhiều thành phần tương tác protein (Wu et al., 2008). Nó cũng đã được báo cáo một số biến thể di truyền trong gen canxi. Các biến thể gây bệnh trong CACNA1C gây ra hội chứng LQT type 8 (Hội chứng Timothy) (Schwartz et al., 2006). Đây là loại hội chứng QT dài không phổ biến, nhưng nó có liên quan đến tỷ lệ tử vong cao nhất. Gần đây, một vài trường hợp hội chứng LQT đã được xác định ở hai gen: CALM1 và CALM2. Hai gen này cùng với mã hóa CALM3 cho protein peaceodulin và các sản phẩm của chúng có trình tự axit amin giống hệt nhau và cả ba gen được biểu hiện ở tâm thất trái của con người (Boczek et al., 2016; Reed et al., 2015). Calmodulin là một protein liên kết Ca + 2 đa chức năng cần thiết cho sự tải nạp tín hiệu Ca + 2 để tác động đến hoạt động của các kênh ion tim, kinase và các protein mục tiêu khác trong tim. Cuối cùng, hội chứng LQT cũng đã được liên kết với ANK2, một loại protein canxi liên quan (Mohler et al., 2003). Gen này mã hóa cho protein Ankyrin-B. Ankyrin là các protein bộ điều hợp liên kết các protein màng, chất vận chuyển và các phân tử kết dính tế bào với tế bào. Biến thể trong ANK2 gây ra sự trao đổi Na + / Ca + 2 và rối loạn chức năng Na + / K + ATPase tạo ra hậu khử cực sớm và trễ của tế nào (EAD và DADs) để đáp ứng với catecholamine
- Hội chứng QT ngắn
Hội chứng QT ngắn (SQTS) là một thực thể gây chết người rất hiếm được mô tả vào năm 2000 (Gussak và cộng sự., 2000). Nó được đặc trưng bằng khoảng QT ngắn (QTc <330 ms), với T cao nhọn và khoảng ngắn giữa đỉnh và kết thúc của sóng T trong ECG. Mức độ nghiêm trọng của các biểu hiện lâm sàng của SQT rất khác nhau, từ không triệu chứng đến ngất tái phát và SCD. Độ tuổi khởi phát các biểu hiện lâm sàng có thể rất trẻ với các báo cáo về các dạng ác tính gây ra cả SCD ở trẻ sơ sinh (Mazzanti et al., 2014). Ngày nay, không có biện pháp điều trị bằng thuốc nào hiệu quả đã được chứng minh để ngăn ngừa rối loạn nhịp tim và cấy ICD là biện pháp duy nhất cho các trường hợp nguy cơ cao. Quinidine đã được thử nghiệm như một phương pháp điều trị để cố gắng kéo dài QT. Rất ít dữ liệu được báo cáo liên quan đến thực hành gắng sức trong SQTS. Vì các căng thẳng giao cảm (stress adrenergic) không liên quan đến sự phát triển của rối loạn nhịp tim đe dọa tính mạng, cũng như không có môn thể thao cạnh tranh cũng như không có hoạt động thời gian trung bình được có phép đến tận khi có nhiều tư liệu hơn có thể (Ackerman et al., 2015)
Tập trung vào di truyền học, một số lượng hạn chế các biến thể di truyền gây bệnh đã thấy có liên quan với SQTS. Những thay đổi di truyền này đã được xác định ở 6 gen khác nhau (KCNQ1, KCNJ2, KCNH2, CACNA1C, CACNB2 và CACNA2D1), theo mô hình di truyền nhiễm sắc thể trội (Behere và Weindling, 2015). Điều này phải là một sự thâm nhập cao và một phân tích di truyền toàn diện xác định sự thay đổi di truyền ở gần 60% các trường hợp được chẩn đoán lâm sàng. Hội chứng QT ngắn type 1 có liên quan đến các biến thể di truyền trong KCNH2 gây ra sự kích hoạt nhanh dòng kali, với chức năng Ikr tăng cường và điện thế hoạt động thất ngắn lại (Brugada và cộng sự., 2004). Hội chứng QT ngắn type 2 có liên quan đến biến thể di truyền ở KCNQ1, tăng cường chức năng của kênh kali, dẫn đến rút ngắn điện thế hoạt động (Hong và cộng sự, 2005). Hội chứng QT ngắn type 3 gây ra do các biến thể di truyền trong KCNJ2, dẫn đến tăng tốc điện thế hoạt động giai đoạn 3 (Priori và cộng sự, 2005). Các biến thể trong gen canxi cũng đã được báo cáo. Hai trong số các gen này (CACNA1C, CACNB2) được liên kết với BrS và rút ngắn khoảng QT, như đã trình bày ở trên (Antzelevitch và cộng sự, 2007). Gen canxi thứ ba là CACNA2D1 và chỉ có 1 biến thể gây bệnh có liên quan đến SQTS (CM111612) (Templin và cộng sự, 2011).
- Nhịp nhanh thất đa hình liên quan đến Catecholamine
Nhịp tim nhanh thất đa hình liên quan đến catecholamine (Catecholaminergic polymorphic ventricular tachycardia: CPVT) là một thực thể tim hiếm gặp đặc trưng bằng nhịp nhanh thất đa hình hai chiều có đặc tính bằng rối loạn nhịp tim nghiêm trọng ở tim bình thường (Refaat và cộng sự, 2016). Hội chứng này có liên quan đến ECG bình thường khi nghỉ (đôi khi có nhịp tim chậm và sóng U), và chúng được kích hoạt bằng kích thích adrenergic và biểu hiện khi gắng sức, căng thẳng hoặc cảm xúc mạnh. Rối loạn nhịp này xảy ra chủ yếu ở trẻ em và thanh thiếu niên và ngày càng được công nhận là nguyên nhân gây SCD không giải thích được ở người trẻ tuổi, chủ yếu ở nam giới trẻ tuổi (30% ở độ tuổi 40) (Mohamed và cộng sự, 2007). Người ta nhận thấy biến cố này xảy ra ở thời thơ ấu (trước 10 tuổi) nhưng các nghiên cứu gần đây cho thấy biểu hiện đầu tiên có thể xảy ra từ giai đoạn sơ sinh đến 40 tuổi. Chẩn đoán CPVT có thể khó khăn đặc biệt ở trẻ nhỏ. Theo dõi ECG gắng sức và Holter 24 giờ có thể rất hữu ích để loại trừ nhịp tim nhanh hai chiều ở trẻ nhỏ, hoạt động thể chất vì CPVT không thể được chẩn đoán bằng ECG lúc nghỉ hoặc các nghiên cứu về tim mạch khác. Điều trị hàng đầu ở các bệnh nhân CPVT là thuốc chẹn beta, đã làm giảm đáng kể ngất và SCD. Hướng dẫn điều trị dựa trên dữ liệu hạn chế; tuy nhiên, ICD được chỉ định cho bệnh nhân SCD được cứu sống hoặc CPVT trong khi gắng sức và ở thanh thiếu niên có CPVT được kiểm soát không hoàn toàn mặc dù đã dùng thuốc liều cao (Imberti cộng sự, 2016). Phẫu thuật loại bỏ hạch giao cảm tim cũng đã được đề xuất để kiểm soát CPVT ở bệnh nhân trơ với betablockers, mặc dù các rối loạn nhịp giảm sau phẫu thuật này có thể là tạm thời hoặc khởi phát muộn. Thể thao trong rối loạn nhịp này được chống chỉ định, bao gồm cả những bệnh nhân được điều trị bằng thuốc chẹn beta (Ackerman và cộng sự, 2015).
Hiện tại, CPVT được gây ra bằng phân phối canxi nội bào bị suy yếu do gần 200 biến thể di truyền gây bệnh ở 5 gen khác nhau (RyR2, CASQ2, KCNJ2, CALM1 và TRDN). Tất cả các gen này cùng nhau giải thích khoảng 60% trường hợp CPVT. Gen chính liên quan đến CPVT là RyR2, chịu trách nhiệm cho gần 50% tất cả các trường hợp (Laitinen và cộng sự, 2001). Thụ thể ryanodine là một kênh canxi nội bào nằm trong mạng lưới sarcoplasmatic và được kích hoạt bằng dòng canxi nhỏ, do đó cho phép dòng canxi được lưu trữ, rất quan trọng trong việc kích hoạt co bóp cơ tim. Một gen khác liên quan đến CPVT là CASQ2 mã hóa cơ tim thành viên gia đình của protein gia đình trong lưới cơ tương của các tế bào cơ điều hòa nồng độ canxi (calsequestrin) hoạt động như lưu trữ canxi bên trong các tế bào cơ (Lahat et al., 2001). Hai protein liên quan đến canxi cũng đã được báo cáo trong các trường hợp CPVT, Calmodulin (CALM1) (Marsman và cộng sự, 2014) và Triadin (TRDN) (Roux-Buisson và cộng sự., 2012). Cho đến nay, chỉ có hai biến thể di truyền đã được liên kết với CPVT (CM128791 và CM128792) trong CALM1. Gần đây, một mối liên hệ tiềm tàng của gen CALM2 trong các đặc điểm lâm sàng chồng chéo của LQTS và CPVT đã được công bố mặc dù vẫn cần để làm rõ vai trò gây bệnh của chúng (Makita et al., 2014). Cuối cùng, Triadin (TRDN) là một protein màng nguyên phân có chứa một miền xuyên màng duy nhất, liên quan đến việc gắn Calributionestrin vào lưới cơ tương và cho phép cặp chức năng của nó với thụ thể Ryanodine điều chỉnh sự phóng thích canxi của cơ tương. Tập trung vào KCNJ2, một vài biến thể di truyền đã được liên kết với CPVT và nó đã được báo cáo là một biến thể (CM111211) ở một bệnh nhân cho thấy hội chứng Andersen-Tawil và giống với CPVT
KẾT LUẬN
Trong vài năm qua, tim mạch học đã được hưởng lợi nhờ cải thiện trong di truyền chủ yếu được áp dụng để chẩn đoán và phòng ngừa SCD. Tuy nhiên, SCD vẫn là một nguyên nhân quan trọng gây tử vong chủ yếu ở dân số trẻ. Mặc dù một số gen đã được báo cáo trong các bệnh kênh ion, phần lớn các gia đình được chẩn đoán lâm sàng vẫn không có nguyên nhân di truyền của bệnh được công nhận. Ngoài ra, việc xác định sớm các cá nhân có nguy cơ và phân tầng nguy cơ là những thách thức chính của các bệnh rối loạn nhịp tim liên quan đến SCD. Do đó, các nghiên cứu sâu hơn kết hợp với gia đình, bác sĩ lâm sàng và các nhà nghiên cứu cơ bản đòi hỏi làm rõ các điểm này và áp dụng điều trị phòng ngừa mang tính các thể hóa.
Theo timmachhoc
PK Đức Tín
Tin tức liên quan
Điện thoại bàn: (028) 3981 2678
Di động: 0903 839 878 - 0909 384 389